| Most people today are all too familiar with the devastating
effects of HIV, the
human immunodeficiency virus. The virus, which is transmitted by
blood-to-blood contact, may produce no symptoms for years. But typically
within 10 to 15 years it destroys key cells of the immune system and
causes AIDS (acquired immunodeficiency syndrome). Loss of immunity
enables microorganisms that would normally be kept in check to proliferate
uncontrollably and can allow life-threatening cancers to develop. So far
in the U.S. alone, AIDS has killed more than 350,000 people and has become
the principal cause of death among those 24 to 44 years old. Another
750,000 Americans harbor the virus, part of some 30 million who are
affected worldwide.
In the past few years, advances in drug therapy
have enabled a number of patients to cheat death. Sophisticated
combinations of medicines
have diminished the levels of virus in the body and restored immune
function. Those feats have been justifiably well publicized, but findings
less known to the public have also caused a stir of late in the AIDS
research community. Investigators have long wondered why some individuals
escape HIV infection despite being at high risk for it and why certain
people who contract the virus progress to AIDS unusually slowly. For
instance, between 1978 and 1984, before donated blood was screened for
HIV, nearly 12,000 hemophiliacs who received tainted blood products became
infected, but 10 to 25 percent of the recipients evaded the virus. And
about 1 percent of individuals who carry HIV remain relatively healthy,
with few or no symptoms and with adequate immune functioning, for
atypically long spans of 15 years or more.
The recent findings reveal that some people who are partly or fully resistant
to HIV infection owe their good fortune to their genes--or, more
precisely, to possession of a particular variant of a gene involved in
immunologic function. This discovery has already sparked intensive efforts
to translate the new genetic understanding into innovative strategies for
preventing and controlling HIV infection. (We should note that we are
using the term "HIV" to
mean HIV-1, the virus responsible for most AIDS worldwide. Another form,
HIV-2, causes AIDS more slowly and is restricted to certain parts of
Africa; genetic resistance to HIV-2 has not yet been studied.)
Precedents in Animals
The story of how the first HIV-resistance gene was unmasked is one of
excruciatingly slow progress followed by a sudden rush of discoveries. The
two of us and our colleagues at the National Cancer Institute
(NCI) initiated a search for such genes in 1984, just a year after HIV
was found to be the cause of AIDS and three years after the disease was
originally identified.
At the time, our project was a radical undertaking. To explain why
people with equal exposure to HIV could have different fates, most workers
in the 1980s focused on genetic characteristics of the virus (such as
variations in the virulence of different strains) or on nongenetic
"co-factors" that might influence the disease-causing power of the virus
(such as infection of the host by another microbe). And we had little
solid evidence that humans could possess genetic protection from AIDS.
Indeed, certain of our colleagues doubted we would find anything on our
genetic "fishing" expedition, a hunt on which we were wagering
considerable time and resources.
Yet we were not operating blindly. Research in animals had clearly
established that genes often affect the acquisition and development of
infections, especially those caused by retroviruses, the
family that includes HIV. Most genes serve as blueprints for proteins, the
molecules that perform the majority of activities in cells. When a
protein-coding gene is switched on, its sequence of building blocks, or
DNA nucleotides, is used as a guide for stringing together the unique
sequence of amino acids in the specified protein. If the gene is polymorphic--present
in more than one form in a population--its variants, or alleles, may give
rise to protein variants that differ in how well they function in the
body. In mice, specific alleles of more than 30 genes had been shown to
confer resistance to retroviruses. Other animal work had also demonstrated
a genetic component to infectious disease. Inbred mice, rats and livestock
are notoriously sensitive to communicable disorders, mainly because
inbreeding leaves them with a limited repertoire of disease-resistance
alleles. In outbred groups, some fraction of a population is likely to
have an allele that protects against a given pathogen; that allele will
enable its owners to survive an epidemic and perpetuate the group. Because
human populations are genetically diverse, we suspected that they, like
other outbred species, possessed many powerful disease-resistance alleles.
Those alleles, perhaps including defenses against HIV, simply remained to
be discovered.
Further, although few pathogen-resisting alleles had been defined
convincingly in humans, several epidemiological studies had noted a strong
genetic influence on disease susceptibility. For instance, one analysis
showed that if a biological parent of an adoptee died of an infectious
disease before age 50, the adoptee had a markedly increased risk of also
dying from an infection.
Unfortunately, science had provided no simple blueprint for finding
HIV-resistance alleles in humans. We therefore combined knowledge and
techniques from three disparate disciplines: AIDS
epidemiology, human
molecular genetics and population genetics theory.
High-Tech Gene Prospecting
First, we needed a source of genes from the populations of interest to
us, such as individuals at high risk for HIV infection who did or did not
become infected after exposure to the virus. If the two groups differed in
their genetic makeup--in their alleles for specific genes--we would
suspect that the genes displaying the variation influenced susceptibility
to HIV infection.
To obtain human DNA for study, we joined forces with public health
epidemiologists who were trying to track the pattern of the still new
epidemic. As part of that effort, the epidemiologists were enlisting cohorts,
or groups of several hundred individuals, at high risk for HIV
infection--notably, homosexual men, users of intravenous drugs and
hemophiliacs who had received contaminated blood products. These cohorts
were to be monitored for years by physicians, who (with the patients'
permission) would supply blood, tissue samples and case reports to
researchers. As blood was collected, our cell biology team, led by Cheryl
Winkler, carefully produced immortal lines of cultured cells that would
provide an unlimited supply of DNA for genetic testing.
To determine which genes to compare, we took advantage of recent
advances in gene mapping, a set of procedures that pinpoints the location
of genes on chromosomes and determines their nucleotide sequences. More
than 6,000 of the approximately 50,000 to 100,000 genes in human
chromosomes have now been mapped.
Back in 1984 fewer than 1,000 had been found. Nevertheless, to test even
1,000 genes in our AIDS cohorts was an impossible task.
We narrowed the choice by drawing on established knowledge of how
retroviruses behave in their hosts. The host is always an unsuspecting
collaborator in establishing infection and enabling pathogens to spread
through tissues. To enter
cells, all viruses must recognize (bind to) certain proteins encoded
by host genes and displayed on the cell. These proteins normally act as receptors
for other host molecules, but viruses can co-opt the receptors, using them
as springboards for entry into a cell.
Once in a cell, retroviruses insidiously insert their genes into a
host's chromosomes. They thereby ensure that viral genes--which can
direct the synthesis of an endless supply of viral particles--are passed
to each new generation of cells whenever the initial host cell replicates.
Here, again, the viruses require help from the host. They must recruit
several cellular enzymes to splice viral genes into chromosomes, to
produce fresh viral particles and even to evade the host's immune
defenses.
With such understanding to guide us, we originally decided to
concentrate on about 50 genes whose proteins could potentially influence
HIV's life cycle. We also examined 250 polymorphic (variable) DNA segments
that had been identified in chromosomal sites between genes. If our
subjects differed in these segments, those differences would indicate that
alleles of nearby genes might also vary systematically between the groups.
We could then perform a fairly narrow search for those genes and try to
determine their function in cells and their role in HIV infection.
Finally, to pinpoint genetic traits that confer resistance to HIV, we
borrowed strategies from human population genetics. We divided each cohort
into two groups, according to selected aspects of their health--for
example, those infected with HIV versus those who remained free of it
after extensive exposure; infected patients who progressed to AIDS rapidly
versus those who progressed slowly if at all; or infected patients who
acquired a specific AIDS-related disease (such as Pneumocystis
carinii pneumonia or Kaposi's sarcoma)
versus those who did not.
Having made these divisions, we compared how often each known allele or
polymorphic segment appeared in the groups. We also compared what are
called genotypes. An individual inherits two copies of all genes outside
the sex chromosomes (one copy from the mother and one from the father).
The pair of alleles at a particular chromosomal locus, or gene address,
constitutes the genotype. Someone who inherits two identical alleles of a
given gene is said to be a homozygote; someone who inherits two distinct
alleles is said to be a heterozygote. In our screening tests, we noted the
percentage of patients in each group who were homozygous for a known
allele and the percentage of patients that were heterozygous. Appreciable
differences in allele or genotype frequencies, or both, in two subject
groups would indicate that the gene under study probably accounted for the
divergent fates of the subject groups.
For years we continued to add more patients, more genes, more
polymorphic segments and more sophisticated computer programs to analyze
the data. Periodically, we thought we noted genetic differences, but they
nearly always evaporated under close inspection. Meanwhile we monitored
the many advances in understanding of human immunology and in the behavior
of HIV in the body, always seeking ideas for other genes to study. Late in
1995 and early in 1996--more than a decade after we began this massive and
tedious effort--cracks finally appeared in the dike.
Good Clues, at Last
Those cracks were created by other research teams who resolved two
long-standing mysteries relating to HIV's molecular interaction with host
cells. With those solutions came clues to genes involved in resistance to
HIV. By the mid-1990s scientists and nonscientists alike were well aware
that HIV caused immune deficiency mainly by depleting white blood cells
known as T lymphocytes that displayed a protein called CD4 on their
surface. These T cells
normally orchestrate many aspects of the immune response to viruses. It
was also known, albeit less widely, that HIV can infect and persist for
years in another class of CD4-carrying immune cells called macrophages.
HIV does not destroy macrophages and finds a safe haven in them.
The CD4 molecules on T lymphocytes and on macrophages usually
participate in signaling between immune cells. But when HIV enters the
picture, CD4 molecules bind to a sugary protein (gp120) protruding from
HIV's outer envelope and, in so doing, help the virus to gain entry into
the bound cells. Yet experiments had shown that CD4, though necessary for
HIV infiltration of cells, was not sufficient; the cells also had to
display at least one more protein to which the virus could bind. More than
10 years after the discovery of HIV, however, scientists still had no clue
to the nature of that second receptor.
The other puzzle related to a discovery reported in 1986 by Jay A. Levy
of the University of California at San Francisco. He found that a class of
T lymphocytes displaying a different protein--CD8--secreted molecules,
termed suppressive factors, that blocked HIV from invading normally
susceptible cells in culture. Suppressive factors that limited virus
infection had also been shown to exist in African monkeys that harbored
SIV (the simian form of HIV) yet did not advance to AIDS, as well as in
people who survive HIV infection for an unusually long time. The identity
of these sundry suppressive factors remained to be determined, however. In
December 1995 Robert C. Gallo, then at the NCI, and other collaborators
announced that they had identified three related suppressive factors that
could block infection by HIV variants that prefer to colonize macrophages
(so-called M-tropic strains). All three factors turned out to be known chemokines:
short strings of amino acids responsible for luring immune cells to
injured or diseased tissues.
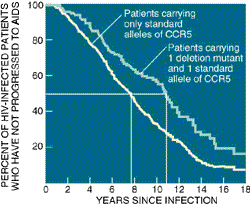 |
| COMPARISON of how long HIV-infected individuals
lived without progressing to AIDS revealed that patients harboring
one deletion mutant of the CCR5 gene (green line) avoided AIDS
longer than patients carrying only standard CCR5 alleles (yellow
line). For instance, it took about 11 years for 50 percent of the
first group to advance to AIDS but about eight years for half of the
second group to reach that point. |
Many investigators still grappling with the first puzzle--the search
for HIV's second receptor--understood that chemokines work their effects
on defensive cells by binding to surface proteins. It seemed possible that
the chemokines isolated by Gallo's group--named RANTES, MIP-1a and
MIP-1b--might interfere with HIV entry into immune cells by binding to and
blocking some cell surface protein that HIV required for access to the
interior. In other words, the cell-surface receptor (or receptors) for
Gallo's chemokines could well lead a double life as the second receptor
for HIV on macrophages and perhaps on other CD4-bearing cells.
The notion defied immediate testing because the cellular receptor for
RANTES and its cousins had not yet been isolated. But discoveries reported
early in 1996 made such tests possible and provided us, and others, with
new genes to screen as resistance factors.
First, Edward A. Berger and his colleagues at the National Institute of Allergy and
Infectious Diseases isolated the second
receptor for HIV variants that prefer to colonize T lymphocytes (T-tropic
strains). It was a chemokine receptor, albeit one (now called CXCR4) that
bound a chemokine distinct from RANTES, MIP-1a and MIP-1b. If Gallo's
findings had not convinced AIDS researchers that chemokine receptors
played a part in HIV infectivity, Berger's results drove the point home.
Almost simultaneously, Michael Samson and Marc Parmentier of the Free
University of Brussels and their collaborators isolated the gene for a
receptor onto which RANTES, MIP-1a and MIP-1b all hook when they draw
defensive cells to damaged tissue. Within two months, five separate groups
proved that the encoded protein, now known as CCR5, was also the elusive
second receptor for M-tropic strains of HIV.
Combined with observations from other studies, the new chemokine
receptor findings critically refined understanding of how HIV infections
become established and progress. HIV initiates infection by setting up
residence primarily in macrophages. It enters these cells by linking its
gp120 protein with two receptors on macrophages: CD4 and CCR5. Once inside
the macrophages, HIV synthesizes large quantities of virus and challenges
the immune system to its limits.
Years later the constantly mutating virus can alter the gene for gp120
in a way that causes the gp120 protein to change its second-receptor
allegiance. The genetic change causes the region that recognizes CCR5 to
bind more effectively to CXCR4 on T lymphocytes. Now the HIV population
becomes dominated by T-tropic variants--those preferring to infect T
cells.
This shift in attraction soon becomes deadly, because T-tropic viruses
kill the cells they infect. Not surprisingly, the shift is often followed
swiftly by an overall drop in CD4 T cell concentrations in patients and,
simultaneously, by the onset of the opportunistic infections and cancers
that for many years defined progression to AIDS. Today the Centers for Disease Control and Prevention
formally defines AIDS by the presence of AIDS-defining illnesses or by a
drop in CD4 T cells to fewer than 200 per cubic millimeter of blood;
normal levels are about 1,000 per cubic millimeter.
The Expedition Succeeds
As soon as we knew that CCR5 and CXCR4 were co-receptors for HIV, we
immediately decided to see whether the genes for those proteins affected
resistance to HIV in our cohorts. To pursue this idea, we had to determine
whether the CCR5 and CXCR4 genes were polymorphic. If everyone had
identical versions of those genes, the genes could not account for
differences in susceptibility to HIV.
All copies of the CXCR4 gene we examined were the same. But in July
1996 Mary Carrington of our group discovered that a major variant of the
normal CCR5 gene occurred in about one in five individuals. Comparisons of
the nucleotide sequences of the two CCR5 alleles revealed that the less
common one was missing 32 nucleotides. Because of the way the genetic code
works, we knew that the loss would result in the premature creation of a
"stop" code in the gene and would, in turn, cause the cells to manufacture
a severely foreshortened version of the CCR5 protein.
When we divided nearly 2,000 high-risk patients into infected and
noninfected groups and compared their CCR5 genotypes, we found dramatic
differences. Some 3 percent of the noninfected individuals carried only
the deletion mutant of CCR5 in their cells (that is, were homozygous for
the mutant). In contrast, not one patient out of 1,343 in the infected
group was homozygous for the deletion mutant. The difference--which
indicated homozygosity for the deletion mutant was protective against
HIV--was highly significant statistically and was certainly no
coincidence. Moreover, the apparent protection provided by having solely
mutant CCR5 alleles did not depend on the route of infection: no
hemophiliacs, homosexuals or drug users who were homozygous contracted
HIV. We suspected that homozygosity for the deletion mutant shielded
patients because they manufactured only truncated CCR5 proteins that
either failed to reach the cell surface or were so deformed that they
could not dock with HIV.
Within a few weeks after submitting a paper on these remarkable
findings to the journal Science,
we learned we were not alone in searching for polymorphisms in chemokine
receptors. Nathaniel
R. Landau and Richard A. Koup of
the Aaron Diamond AIDS
Research Center in New York City and their co-workers had
independently discovered the same 32-base-pair deletion allele. They had
been studying a group of homosexual men who had many high-risk sexual
exposures to HIV but had never become infected.
Examination of white blood cells from two of these men indicated that
the CCR5 protein was absent from the cell surface. A look at the
nucleotide sequence of the CCR5 genes revealed that both men were
homozygous for the deletion mutant. Further, in other work, Samson and
Parmentier's team had searched for and failed to turn up any homozygotes
for the deletion allele in a group of 743 HIV-infected people. (Those two
reports appeared in August 1996, ours in September.)
Subsequent studies uncovered no homozygotes among Africans, Asians or
African-Americans but indicated that 1 to 2 percent of
Caucasian-Americans--those descended from Europeans or western Asians--are
homozygous for the mutation. Further, when we looked at the genotypes of
uninfected people known to have had extremely high exposure to HIV
(through engaging in unsafe sex repeatedly or receiving high doses of
HIV-contaminated clotting factors during treatment for hemophilia), we saw
that as many as 20 percent of these individuals were homozygous for the
deletion mutant. Resistance to infection in the other 80 percent must have
come from other genetic or nongenetic sources.
It stood to reason that if two mutant CCR5 genes provided complete
protection from HIV, possession of one mutant and one normal allele might
provide partial protection, by halving the number of functional CCR5
proteins made by a cell. When we analyzed the time between infection and
the appearance of AIDS-defining diseases, we found that the onset of overt
AIDS was postponed for two to three years in HIV-infected individuals who
carried one deletion allele. This delay was apparent both in homosexual
men and in hemophiliacs. This heterozygous genotype (which occurs in
approximately 20 percent of Caucasian-Americans) also delayed the time at
which CD4 T cell levels fell below 200 per cubic millimeter of blood.
The excitement was overwhelming. The deletion mutant, when inherited
from both parents, did indeed appear to provide powerful genetic
protection against HIV even after repeated exposures. And inheritance of a
single deletion mutant could slow progression to AIDS in infected
individuals. These results implied that treatments able to block the
interaction of HIV with the normal CCR5 protein might help protect healthy
people from HIV infection or delay the advance to AIDS in people who have
already contracted the virus.
For years, pharmaceutical companies had focused their anti-HIV
therapeutic efforts on the virus alone, giving little attention to how the
host's cellular machinery collaborates in establishing chronic disease.
The drugs used in combination therapy, for example, interfere directly
with the activities of HIV itself, such as by preventing certain of its
enzymes from functioning. The new genetic results suggested that targeting
the host's complicity in the progression to AIDS could open previously
unimagined avenues for controlling HIV replication in infected patients or
for preventing HIV infection in the first place.
Implications for Treatment
Not surprisingly, many investigators quickly began considering ways to
keep HIV and the CCR5 protein from interacting. In theory, such strategies
could involve substances that sheathe gp120. In practice, however, most
efforts are searching for ways to plug the HIV binding site on CCR5.
An initial concern was that blocking CCR5 would be dangerous--that it
might impair immunity by making macrophages deaf to the call of RANTES and
related chemokines. But that worry was soon allayed. Individuals who
possess two mutant alleles have no obvious immune dysfunction or tissue
pathology and appear to be quite healthy. Evidently, other chemokine
receptors can compensate for the lack of CCR5. Two of them (CCR2B and
CCR3) can also serve as co-receptors for HIV, although they generally do
not perform that nefarious job nearly as effectively as CCR5.
Among the therapeutic strategies under consideration is direct delivery
of molecules that would obstruct CCR5's binding site for HIV. Such
molecules could include chemokines or synthetic derivatives of chemokines.
For instance, an international team of investigators has developed a
modified chemical derivative of RANTES that shows promise in test-tube
studies.
Other molecular "plugs" could include synthetic antibodies--larger
immune molecules that would specifically home to CCR5 and bar attachment
by HIV. Additional approaches involve vaccinating people with fragments of
CCR5 that could induce the recipient's immune system to produce its own
CCR5-binding antibodies. Alternatively, researchers could use genetic
engineering to provide macrophages with new genes whose products would
prevent CCR5 from being made or would stop CCR5 from serving as a docking
site for HIV. For some patients facing imminent death--such as those in
the final stage of AIDS who also have lymphoma--our group is considering
modifying a radical treatment increasingly applied to advanced cases of
blood or breast cancers. When curing these cancers is the aim, patients
are given extremely high doses of chemotherapy or radiation to eradicate
all cancer cells. Because that therapy destroys the blood-producing cells
of the bone marrow (including the ones that give rise to the immune
system), physicians then reconstitute the patient's immune system by
delivering healthy, tissue-compatible marrow.
In the case of AIDS patients, we would aim to destroy all HIV-infected
blood cells and then to rescue the patient with bone marrow from donors
who are homozygous for the deletion mutant of the CCR5 gene. This last
step would, we hope, help protect the patient from new HIV infection and
also help prevent the cell-to-cell spread of any HIV particles that
somehow survived the HIV-destroying therapy.
The idea of simultaneously curing patients and giving them protection
from residual or sequestered HIV holds great appeal, but we are
approaching bone marrow therapy cautiously because of a few important
concerns. For one thing, bone marrow transplants are inherently risky:
immunologic differences between the donor and the recipient can cause
rejection of the transplant or, worse, can cause the immune cells in the
donor marrow to attack the tissues of the host and kill the patient.
In addition, in recent months, a few individuals have surfaced who are
homozygous for the deletion mutant but who have nonetheless become
infected with HIV. We do not yet know how the infection became
established, but some signs indicate that these rare patients met with an
unusual "hot," or highly virulent, T-tropic strain of the type that
typically emerges only in the late stages of HIV infection.
Until now, conventional wisdom held that T-tropic viruses were unable
to spread infection from one person to another. They seemed to be
recognized and destroyed by the healthy immune system of newly exposed
individuals. Successful infection was thought to require M-tropic viruses,
which quietly multiplied to high levels in macrophages without eliciting
destruction of those cells. Some evidence suggests that the patients who
became infected even though they were homozygous for the deletion mutant
were merely unlucky and simply encountered odd T-tropic strains that were
able to circumvent immune defenses and establish infection without needing
M-tropic strains to lay the groundwork. It is also possible, however, that
the patients' innate resistance to M-tropic strains somehow sped up the
transition of M-tropic strains to hot T-tropic types able to establish
infection on their own.
If CCR5-mediated resistance to M-tropic strains actually encouraged HIV
to turn hot, the finding would mean that bone marrow transplants--and, in
fact, any preventives or therapies aimed at blocking HIV's access to
CCR5--could backfire and encourage, instead of forestall, infection and
advancement to AIDS. The fact that most people who are homozygous for the
deletion allele avoid HIV infection instead of succumbing to severe
T-tropic viruses is reassuring. Nevertheless, before physicians can
routinely treat patients with antagonists of CCR5, investigators need to
show that such drugs improve, rather than diminish, the likelihood of
survival.
As scientists explore safe, effective ways to capitalize on the recent
genetic findings, they also continue to look for other genetic factors
that could suggest additional ways to shield people from AIDS. Indeed, our
group has recently identified a variant of the CCR2B gene that even in a
single copy delays the onset of AIDS by two or three years, just as
heterozygosity for CCR5 does. And earlier this year Jianglin He of
the Dana-Farber Cancer Institute and
his colleagues reported that the CCR3 protein promotes HIV entry into
microglia (immune cells in the brain) and that blockade of the receptor
prevents HIV infection of microglia in the laboratory.
After more than a decade of searching for genetic traits that provide
protection from AIDS, we are indeed pleased by the quickening pace of
discovery. But the main goal must be transforming genetic insights into
novel ways to evade or attack HIV, a virus clever enough to destroy the
very cells meant to eradicate it. Although therapeutic applications remain
speculative for now, we are hopeful that the combined talents of
researchers from many fields will provide a scientific recipe for
reversing the deliberate progression of the AIDS epidemic.
Further Reading
HIV-1 ENTRY COFACTOR: FUNCTIONAL CDNA CLONING OF
SEVEN-TRANSMEMBRANE, G PROTEIN-COUPLED RECEPTOR. Y. Feng, C. C.
Broder, P. E. Kennedy and E. A. Berger in Science, Vol. 272,
pages 872-877; May 10, 1996.
HOMOZYGOUS DEFECT IN HIV-1 CORECEPTOR ACCOUNTS FOR
RESISTANCE OF SOME MULTIPLY-EXPOSED INDIVIDUALS TO HIV-1 INFECTION.
Rong Liu et al. in Cell, Vol. 86, No. 3, pages 367-377; August 9,
1996.
GENETIC RESTRICTION OF HIV-1 INFECTION AND PROGRESSION TO
AIDS BY A DELETION ALLELE OF THE CKR5 STRUCTURAL GENE. Michael Dean
et al. in Science, Vol. 273,
pages 1856-1862; September 27, 1996.
CONTRASTING GENETIC INFLUENCE OF CCR2 AND CCR5 VARIANTS
ON HIV-1 INFECTION AND DISEASE PROGRESSION. Michael W. Smith et al.
in Science (in
press).
Classic Papers in
Genetics in Adobe Acrobat 2.0 PDF format
HIV CAUSES AIDS: KOCHE'S POSTULATES FULFILLED. .
Stephen J. O'Brien and James Goedert in Current Opinion in Immunology
, Vol. 8, pages 613-618; 1996.
IDENTIFICATION OF RANGES, MIP-1a [this is an alpha] AND
MIP-1B AS THE MAJOR HIV-SUPPRESSIVE FACTORS PRODUCED BY CD8+ T CELLS.
. F. Cocchi, A.L. DeVico, A. Garzino-Demo, S.K. Arya, R.C. Gallo
and P. Lusso in Science, Vol. 270,
pages 1811-1815; December 15, 1995.
Related Links
AIDS Information from
The NAMES Project Foundation
Scientists
Confirm Natural Resistance to HIV-1
Classic
Papers in Genetics in Adobe Acrobat 2.0 PDF format
NIAID Researchers
Identify Second Fusion Cofactor for HIV
RRM
Analysis Of The CC-CKR5 As A Co-receptor For HIV-1
A
Gene That Walls Off HIV
NIAID
AIDS Agenda NIAID Researchers Identify Cofactors for Entry of HIV into
Cells
Cells Alive! Human
Immunodeficiency Virus (HIV) Infection
Glossary of
HIV/AIDS Terms
Exhibit:
Cracking Open AIDS's Shell from Scientific American
Exploration:
AIDS Moonshot? from Scientific American
The Authors
STEPHEN
J. O'BRIEN and MICHAEL
DEAN have collaborated for more than a decade. O'Brien has been chief
of the Laboratory of Genomic Diversity at the National Cancer Institute
since 1986. He is internationally recognized for his contributions in
human and animal genetics, evolutionary biology, retrovirology and species
conservation. With two other colleagues, O'Brien also founded and
co-directs NOAHS, a Smithsonian Institution consortium of scientists and
apprentices who apply biotechnology on behalf of species conservation.
Dean is chief of the human genetics section of the Laboratory
of Genomic Diversity, where he applies new genetic techniques to the
study of complex human diseases. The authors dedicate this article to the
memory of Daniel O'Brien, Stephen O'Brien's brother, who died from AIDS in
1994. |